aiida.restapi.translator.data package¶
-
class
aiida.restapi.translator.data.DataTranslator(Class=None, **kwargs)[source]¶ Bases:
aiida.restapi.translator.node.NodeTranslatorTranslator relative to resource ‘data’ and aiida class ~aiida.orm.data.Data
-
__label__= 'data'¶
-
__module__= 'aiida.restapi.translator.data'¶
-
_aiida_class¶ alias of
aiida.orm.data.Data
-
_aiida_type= 'data.Data'¶
-
_qb_type= 'data.Data.'¶
-
_result_type= 'data'¶
-
Submodules¶
-
class
aiida.restapi.translator.data.bands.BandsDataTranslator(**kwargs)[source]¶ Bases:
aiida.restapi.translator.data.DataTranslatorTranslator relative to resource ‘bands’ and aiida class BandsData
-
class
BandsData(*args, **kwargs)¶ Bases:
aiida.orm.data.array.kpoints.KpointsDataClass to handle bands data
-
__abstractmethods__= frozenset([])¶
-
__module__= 'aiida.orm.data.array.bands'¶
-
_abc_cache= <_weakrefset.WeakSet object>¶
-
_abc_negative_cache= <_weakrefset.WeakSet object>¶
-
_abc_negative_cache_version= 119¶
-
_abc_registry= <_weakrefset.WeakSet object>¶
-
_custom_export_format_replacements= {'dat': 'dat_multicolumn', 'gnu': 'gnuplot', 'pdf': 'mpl_pdf', 'png': 'mpl_png', 'py': 'mpl_singlefile'}¶
-
_get_band_segments(cartesian)¶
-
_get_bandplot_data(cartesian, prettify_format=None, join_symbol=None, get_segments=False, y_origin=0.0)¶ Get data to plot a band structure
Parameters: - cartesian – if True, distances (for the x-axis) are computed in cartesian coordinates, otherwise they are computed in reciprocal coordinates. cartesian=True will fail if no cell has been set.
- prettify_format – by default, strings are not prettified. If you want to prettify them, pass a valid prettify_format string (see valid options in the docstring of :py:func:prettify_labels).
- join_symbols – by default, strings are not joined. If you pass a string,
this is used to join strings that are much closer than a given threshold.
The most typical string is the pipe symbol:
|. - get_segments – if True, also computes the band split into segments
- y_origin – if present, shift bands so to set the value specified at
y=0
Returns: a plot_info dictiorary, whose keys are
x(array of distances for the x axis of the plot);y(array of bands),labels(list of tuples in the format (float x value of the label, label string),band_type_idx(array containing an index for each band: if there is only one spin, then it’s an array of zeros, of length equal to the number of bands at each point; if there are two spins, then it’s an array of zeros or ones depending on the type of spin; the length is always equalt to the total number of bands per kpoint).
-
_logger= <celery.utils.log.ProcessAwareLogger object>¶
-
_matplotlib_get_dict(main_file_name='', comments=True, title='', legend=None, legend2=None, y_max_lim=None, y_min_lim=None, y_origin=0.0, prettify_format=None, **kwargs)¶ Prepare the data to send to the python-matplotlib plotting script.
Parameters: - comments – if True, print comments (if it makes sense for the given format)
- plot_info – a dictionary
- setnumber_offset – an offset to be applied to all set numbers (i.e. s0 is replaced by s[offset], s1 by s[offset+1], etc.)
- color_number – the color number for lines, symbols, error bars and filling (should be less than the parameter max_num_agr_colors defined below)
- title – the title
- legend – the legend (applied only to the first of the set)
- legend2 – the legend for second-type spins (applied only to the first of the set)
- y_max_lim – the maximum on the y axis (if None, put the maximum of the bands)
- y_min_lim – the minimum on the y axis (if None, put the minimum of the bands)
- y_origin – the new origin of the y axis -> all bands are replaced by bands-y_origin
- prettify_format – if None, use the default prettify format. Otherwise specify a string with the prettifier to use.
- kwargs – additional customization variables; only a subset is accepted, see internal variable ‘valid_additional_keywords
-
_plugin_type_string= 'data.array.bands.BandsData.'¶
-
_prepare_agr(main_file_name='', comments=True, setnumber_offset=0, color_number=1, color_number2=2, legend='', title='', y_max_lim=None, y_min_lim=None, y_origin=0.0, prettify_format=None)¶ Prepare an xmgrace agr file.
Parameters: - comments – if True, print comments (if it makes sense for the given format)
- plot_info – a dictionary
- setnumber_offset – an offset to be applied to all set numbers (i.e. s0 is replaced by s[offset], s1 by s[offset+1], etc.)
- color_number – the color number for lines, symbols, error bars and filling (should be less than the parameter max_num_agr_colors defined below)
- color_number2 – the color number for lines, symbols, error bars and filling for the second-type spins (should be less than the parameter max_num_agr_colors defined below)
- legend – the legend (applied only to the first set)
- title – the title
- y_max_lim – the maximum on the y axis (if None, put the
maximum of the bands); applied after shifting the origin
by
y_origin - y_min_lim – the minimum on the y axis (if None, put the
minimum of the bands); applied after shifting the origin
by
y_origin - y_origin – the new origin of the y axis -> all bands are replaced by bands-y_origin
- prettify_format – if None, use the default prettify format. Otherwise specify a string with the prettifier to use.
-
_prepare_agr_batch(main_file_name='', comments=True, prettify_format=None)¶ Prepare two files, data and batch, to be plot with xmgrace as: xmgrace -batch file.dat
Parameters: - main_file_name – if the user asks to write the main content on a file, this contains the filename. This should be used to infer a good filename for the additional files. In this case, we remove the extension, and add ‘_data.dat’
- comments – if True, print comments (if it makes sense for the given format)
- prettify_format – if None, use the default prettify format. Otherwise specify a string with the prettifier to use.
-
_prepare_dat_1(*args, **kwargs)¶ Output data in .dat format, using multiple columns for all y values associated to the same x.
Deprecated since version 0.8.1: Use ‘dat_multicolumn’ format instead
-
_prepare_dat_2(*args, **kwargs)¶ Output data in .dat format, using blocks.
Deprecated since version 0.8.1: Use ‘dat_block’ format instead
-
_prepare_dat_blocks(main_file_name='', comments=True)¶ Format suitable for gnuplot using blocks. Columns with x and y (path and band energy). Several blocks, separated by two empty lines, one per energy band.
Parameters: comments – if True, print comments (if it makes sense for the given format)
-
_prepare_dat_multicolumn(main_file_name='', comments=True)¶ Write an N x M matrix. First column is the distance between kpoints, The other columns are the bands. Header contains number of kpoints and the number of bands (commented).
Parameters: comments – if True, print comments (if it makes sense for the given format)
-
_prepare_gnuplot(main_file_name='', title='', comments=True, prettify_format=None, y_max_lim=None, y_min_lim=None, y_origin=0.0)¶ Prepare an gnuplot script to plot the bands, with the .dat file returned as an independent file.
Parameters: - main_file_name – if the user asks to write the main content on a file, this contains the filename. This should be used to infer a good filename for the additional files. In this case, we remove the extension, and add ‘_data.dat’
- title – if specified, add a title to the plot
- comments – if True, print comments (if it makes sense for the given format)
- prettify_format – if None, use the default prettify format. Otherwise specify a string with the prettifier to use.
-
_prepare_json(main_file_name='', comments=True)¶ Prepare a json file in a format compatible with the AiiDA band visualizer
Parameters: comments – if True, print comments (if it makes sense for the given format)
-
_prepare_mpl_pdf(main_file_name='', *args, **kwargs)¶ Prepare a python script using matplotlib to plot the bands, with the JSON returned as an independent file.
For the possible parameters, see documentation of
_matplotlib_get_dict()
-
_prepare_mpl_png(main_file_name='', *args, **kwargs)¶ Prepare a python script using matplotlib to plot the bands, with the JSON returned as an independent file.
For the possible parameters, see documentation of
_matplotlib_get_dict()
-
_prepare_mpl_singlefile(*args, **kwargs)¶ Prepare a python script using matplotlib to plot the bands
For the possible parameters, see documentation of
_matplotlib_get_dict()
-
_prepare_mpl_withjson(main_file_name='', *args, **kwargs)¶ Prepare a python script using matplotlib to plot the bands, with the JSON returned as an independent file.
For the possible parameters, see documentation of
_matplotlib_get_dict()
-
_query_type_string= 'data.array.bands.'¶
-
_set_pbc(value)¶ validate the pbc, then store them
-
_validate_bands_occupations(bands, occupations=None, labels=None)¶ Validate the list of bands and of occupations before storage. Kpoints must be set in advance. Bands and occupations must be convertible into arrays of Nkpoints x Nbands floats or Nspins x Nkpoints x Nbands; Nkpoints must correspond to the number of kpoints.
-
array_labels¶ Get the labels associated with the band arrays
-
get_bands(also_occupations=False, also_labels=False)¶ Returns an array (nkpoints x num_bands or nspins x nkpoints x num_bands) of energies. :param also_occupations: if True, returns also the occupations array. Default = False
-
set_bands(bands, units=None, occupations=None, labels=None)¶ Set an array of band energies of dimension (nkpoints x nbands). Kpoints must be set in advance. Can contain floats or None. :param bands: a list of nkpoints lists of nbands bands, or a 2D array of shape (nkpoints x nbands), with band energies for each kpoint :param units: optional, energy units :param occupations: optional, a 2D list or array of floats of same shape as bands, with the occupation associated to each band
-
set_kpointsdata(kpointsdata)¶ Load the kpoints from a kpoint object. :param kpointsdata: an instance of KpointsData class
-
show_mpl(**kwargs)¶ Call a show() command for the band structure using matplotlib. This uses internally the ‘mpl_singlefile’ format, with empty main_file_name.
Other kwargs are passed to self._exportstring.
-
units¶ Units in which the data in bands were stored. A string
-
-
__label__= 'bands'¶
-
__module__= 'aiida.restapi.translator.data.bands'¶
-
_aiida_class¶ alias of
aiida.orm.data.array.bands.BandsData
-
_aiida_type= 'data.array.bands.BandsData'¶
-
_qb_type= 'data.array.bands.BandsData.'¶
-
_result_type= 'bands'¶
-
class
Translator for CifData
-
class
aiida.restapi.translator.data.cif.CifDataTranslator(**kwargs)[source]¶ Bases:
aiida.restapi.translator.data.DataTranslatorTranslator relative to resource ‘structures’ and aiida class CifData
-
class
CifData(**kwargs)¶ Bases:
aiida.orm.data.singlefile.SinglefileDataWrapper for Crystallographic Interchange File (CIF)
Note
the file (physical) is held as the authoritative source of information, so all conversions are done through the physical file: when setting
aseorvalues, a physical CIF file is generated first, the values are updated from the physical CIF file.-
__abstractmethods__= frozenset([])¶
-
__init__(**kwargs)¶ Initialises an instance of CifData.
-
__module__= 'aiida.orm.data.cif'¶
-
_abc_cache= <_weakrefset.WeakSet object>¶
-
_abc_negative_cache= <_weakrefset.WeakSet object>¶
-
_abc_negative_cache_version= 94¶
-
_abc_registry= <_weakrefset.WeakSet object>¶
-
_get_aiida_structure(converter='ase', store=False, **kwargs)¶ Creates
aiida.orm.data.structure.StructureData.Parameters: - converter – specify the converter. Default ‘ase’.
- store – if True, intermediate calculation gets stored in the AiiDA database for record. Default False.
- primitive_cell – if True, primitive cell is returned, conventional cell if False. Default False.
Returns:
-
_get_object_ase()¶ Converts CifData to ase.Atoms
Returns: an ase.Atoms object
-
_get_object_pycifrw()¶ Converts CifData to PyCIFRW.CifFile
Returns: a PyCIFRW.CifFile object
-
_logger= <celery.utils.log.ProcessAwareLogger object>¶
-
_parse_policies= ['eager', 'lazy']¶
-
_plugin_type_string= 'data.cif.CifData.'¶
-
_prepare_cif(main_file_name='')¶ Return CIF string of CifData object.
If parsed values are present, a CIF string is created and written to file. If no parsed values are present, the CIF string is read from file.
-
_prepare_tcod(main_file_name='', **kwargs)¶ Write the given CIF to a string of format TCOD CIF.
-
_query_type_string= 'data.cif.'¶
-
_scan_types= ['standard', 'flex']¶
-
_set_defaults¶ Add defaults for some attributes.
-
_set_incompatibilities= [('ase', 'file'), ('ase', 'values'), ('file', 'values')]¶
-
_validate()¶ Validates MD5 hash of CIF file.
-
ase¶ ASE object, representing the CIF.
Note
requires ASE module.
-
classmethod
from_md5(md5)¶ Return a list of all CIF files that match a given MD5 hash.
Note
the hash has to be stored in a
_md5attribute, otherwise the CIF file will not be found.
-
generate_md5()¶ Computes and returns MD5 hash of the CIF file.
-
get_ase(**kwargs)¶ Returns ASE object, representing the CIF. This function differs from the property
aseby the possibility to pass the keyworded arguments (kwargs) to ase.io.cif.read_cif().Note
requires ASE module.
-
get_formulae(mode='sum')¶ Return chemical formulae specified in CIF file.
Note: This does not compute the formula, it only reads it from the appropriate tag. Use refine_inline to compute formulae.
-
classmethod
get_or_create(filename, use_first=False, store_cif=True)¶ Pass the same parameter of the init; if a file with the same md5 is found, that CifData is returned.
Parameters: - filename – an absolute filename on disk
- use_first – if False (default), raise an exception if more than one CIF file is found. If it is True, instead, use the first available CIF file.
- store_cif (bool) – If false, the CifData objects are not stored in the database. default=True.
Return (cif, created): where cif is the CifData object, and create is either True if the object was created, or False if the object was retrieved from the DB.
-
get_spacegroup_numbers()¶ Get the spacegroup international number.
-
has_attached_hydrogens()¶ Check if there are hydrogens without coordinates, specified as attached to the atoms of the structure. :return: True if there are attached hydrogens, False otherwise.
-
has_partial_occupancies()¶ Check if there are float values in the atom occupancies. :return: True if there are partial occupancies, False otherwise.
-
parse(scan_type=None)¶ Parses CIF file and sets attributes.
Parameters: scan_type – See set_scan_type
-
static
read_cif(index=-1, **kwargs)¶ A wrapper method that simulates the behavior of the old function ase.io.cif.read_cif by using the new generic ase.io.read function.
-
set_ase(aseatoms)¶
-
set_file(filename)¶ Set the file.
If the source is set and the MD5 checksum of new file is different from the source, the source has to be deleted.
-
set_parse_policy(parse_policy)¶ Set the parse policy.
Parameters: parse_policy – Either ‘eager’ (parse CIF file on set_file) or ‘lazy’ (defer parsing until needed)
-
set_scan_type(scan_type)¶ Set the scan_type for PyCifRW.
The ‘flex’ scan_type of PyCifRW is faster for large CIF files but does not yet support the CIF2 format as of 02/2018. See the CifFile.ReadCif function
Parameters: scan_type – Either ‘standard’ or ‘flex’ (see _scan_types)
-
set_values(values)¶ Set internal representation to values.
Warning: This also writes a new CIF file.
Parameters: values – PyCifRW CifFile object Note
requires PyCifRW module.
-
store(*args, **kwargs)¶ Store the node.
-
values¶ PyCifRW structure, representing the CIF datablocks.
Note
requires PyCifRW module.
-
-
__label__= 'cifs'¶
-
__module__= 'aiida.restapi.translator.data.cif'¶
-
_aiida_class¶ alias of
aiida.orm.data.cif.CifData
-
_aiida_type= 'data.cif.CifData'¶
-
_qb_type= 'data.cif.CifData.'¶
-
_result_type= 'cifs'¶
-
class
-
class
aiida.restapi.translator.data.kpoints.KpointsDataTranslator(**kwargs)[source]¶ Bases:
aiida.restapi.translator.data.DataTranslatorTranslator relative to resource ‘kpoints’ and aiida class KpointsData
-
class
KpointsData(*args, **kwargs)¶ Bases:
aiida.orm.data.array.ArrayDataClass to handle array of kpoints in the Brillouin zone. Provide methods to generate either user-defined k-points or path of k-points along symmetry lines. Internally, all k-points are defined in terms of crystal (fractional) coordinates. Cell and lattice vector coordinates are in Angstroms, reciprocal lattice vectors in Angstrom^-1 . :note: The methods setting and using the Bravais lattice info assume the PRIMITIVE unit cell is provided in input to the set_cell or set_cell_from_structure methods.
-
__abstractmethods__= frozenset([])¶
-
__init__(*args, **kwargs)¶ Initialize the object Node.
Parameters: uuid – if present, the Node with given uuid is loaded from the database. (It is not possible to assign a uuid to a new Node.)
-
__module__= 'aiida.orm.data.array.kpoints'¶
-
_abc_cache= <_weakrefset.WeakSet object>¶
-
_abc_negative_cache= <_weakrefset.WeakSet object>¶
-
_abc_negative_cache_version= 94¶
-
_abc_registry= <_weakrefset.WeakSet object>¶
-
_change_reference(kpoints, to_cartesian=True)¶ Change reference system, from cartesian to crystal coordinates (units of b1,b2,b3) or viceversa. :param kpoints: a list of (3) point coordinates :return kpoints: a list of (3) point coordinates in the new reference
-
_dimension¶ Dimensionality of the structure, found from its pbc (i.e. 1 if it’s a 1D structure, 2 if its 2D, 3 if it’s 3D …). :return dimensionality: 0, 1, 2 or 3 :note: will return 3 if pbc has not been set beforehand
-
_find_bravais_info(epsilon_length=1e-05, epsilon_angle=1e-05)¶ Finds the Bravais lattice of the cell passed in input to the Kpoint class :note: We assume that the cell given by the cell property is the primitive unit cell.
Deprecated since version 0.11: Use the methods inside the aiida.tools.data.array.kpoints module instead.
Returns: a dictionary, with keys short_name, extended_name, index (index of the Bravais lattice), and sometimes variation (name of the variation of the Bravais lattice) and extra (a dictionary with extra parameters used by the get_special_points method)
-
_get_or_create_bravais_lattice(epsilon_length=1e-05, epsilon_angle=1e-05)¶ Try to get the bravais_lattice info if stored already, otherwise analyze the cell with the default settings and save this in the attribute.
Deprecated since version 0.11: Use the methods inside the aiida.tools.data.array.kpoints module instead.
Parameters: - epsilon_length – threshold on lengths comparison, used to get the bravais lattice info
- epsilon_angle – threshold on angles comparison, used to get the bravais lattice info
Return bravais_lattice: the dictionary containing the symmetry info
-
_logger= <celery.utils.log.ProcessAwareLogger object>¶
-
_plugin_type_string= 'data.array.kpoints.KpointsData.'¶
-
_query_type_string= 'data.array.kpoints.'¶
-
_set_bravais_lattice(value)¶ Validating function to set the bravais_lattice dictionary
Deprecated since version 0.11.
-
_set_cell(value)¶ Validate if ‘value’ is a allowed crystal unit cell :param value: something compatible with a 3x3 tuple of floats
-
_set_labels(value)¶ set label names. Must pass in input a list like:
[[0,'X'],[34,'L'],... ]
-
_set_pbc(value)¶ validate the pbc, then store them
-
_set_reciprocal_cell()¶ Sets the reciprocal cell in units of 1/Angstrom from the internally set cell
-
_validate_kpoints_weights(kpoints, weights)¶ Validate the list of kpoints and of weights before storage. Kpoints and weights must be convertible respectively to an array of N x dimension and N floats
-
bravais_lattice¶ The dictionary containing informations about the cell symmetry
Deprecated since version 0.11.
-
cell¶ The crystal unit cell. Rows are the crystal vectors in Angstroms. :return: a 3x3 numpy.array
-
find_bravais_lattice(epsilon_length=1e-05, epsilon_angle=1e-05)¶ Analyze the symmetry of the cell. Allows to relax or tighten the thresholds used to compare angles and lengths of the cell. Save the information of the cell used for later use (like getting special points). It has to be used if the user wants to be sure the right symmetries are recognized. Otherwise, this function is automatically called with the default values.
If the right symmetry is not found, be sure also you are providing cells with enough digits.
If node is already stored, just returns the symmetry found before storing (if any).
Deprecated since version 0.11: Use the methods inside the aiida.tools.data.array.kpoints module instead.
Return (str) lattice_name: the name of the bravais lattice and its eventual variation
-
get_desc()¶ Returns a string with infos retrieved from kpoints node’s properties. :param node: :return: retstr
-
get_kpoints(also_weights=False, cartesian=False)¶ Return the list of kpoints
Parameters: - also_weights – if True, returns also the list of weights. Default = False
- cartesian – if True, returns points in cartesian coordinates, otherwise, returns in crystal coordinates. Default = False.
-
get_kpoints_mesh(print_list=False)¶ Get the mesh of kpoints.
Parameters: print_list – default=False. If True, prints the mesh of kpoints as a list Raises: AttributeError – if no mesh has been set Return mesh,offset: (if print_list=False) a list of 3 integers and a list of three floats 0<x<1, representing the mesh and the offset of kpoints Return kpoints: (if print_list = True) an explicit list of kpoints coordinates, similar to what returned by get_kpoints()
-
get_special_points(cartesian=False, epsilon_length=1e-05, epsilon_angle=1e-05)¶ Get the special point and path of a given structure.
References:
- In 2D, coordinates are based on the paper: R. Ramirez and M. C. Bohm, Int. J. Quant. Chem., XXX, pp. 391-411 (1986)
- In 3D, coordinates are based on the paper: W. Setyawan, S. Curtarolo, Comp. Mat. Sci. 49, 299 (2010)
Deprecated since version 0.11: Use the methods inside the aiida.tools.data.array.kpoints module instead.
Parameters: - cartesian – If true, returns points in cartesian coordinates. Crystal coordinates otherwise. Default=False
- epsilon_length – threshold on lengths comparison, used to get the bravais lattice info
- epsilon_angle – threshold on angles comparison, used to get the bravais lattice info
Returns point_coords: a dictionary of point_name:point_coords key,values.
Returns path: the suggested path which goes through all high symmetry lines. A list of lists for all path segments. e.g. [(‘G’,’X’),(‘X’,’M’),…] It’s not necessarily a continuous line.
Note: We assume that the cell given by the cell property is the primitive unit cell
-
labels¶ Labels associated with the list of kpoints. List of tuples with kpoint index and kpoint name:
[(0,'G'),(13,'M'),...]
-
pbc¶ The periodic boundary conditions along the vectors a1,a2,a3.
Returns: a tuple of three booleans, each one tells if there are periodic boundary conditions for the i-th real-space direction (i=1,2,3)
-
set_cell(cell, pbc=None)¶ Set a cell to be used for symmetry analysis. To set a cell from an AiiDA structure, use “set_cell_from_structure”.
Parameters: - cell – 3x3 matrix of cell vectors. Orientation: each row represent a lattice vector. Units are Angstroms.
- pbc – list of 3 booleans, True if in the nth crystal direction the structure is periodic. Default = [True,True,True]
-
set_cell_from_structure(structuredata)¶ Set a cell to be used for symmetry analysis from an AiiDA structure. Inherits both the cell and the pbc’s. To set manually a cell, use “set_cell”
Parameters: structuredata – an instance of StructureData
-
set_kpoints(kpoints, cartesian=False, labels=None, weights=None, fill_values=0)¶ Set the list of kpoints. If a mesh has already been stored, raise a ModificationNotAllowed
Parameters: - kpoints –
a list of kpoints, each kpoint being a list of one, two or three coordinates, depending on self.pbc: if structure is 1D (only one True in self.pbc) one allows singletons or scalars for each k-point, if it’s 2D it can be a length-2 list, and in all cases it can be a length-3 list. Examples:
- [[0.,0.,0.],[0.1,0.1,0.1],…] for 1D, 2D or 3D
- [[0.,0.],[0.1,0.1,],…] for 1D or 2D
- [[0.],[0.1],…] for 1D
- [0., 0.1, …] for 1D (list of scalars)
For 0D (all pbc are False), the list can be any of the above or empty - then only Gamma point is set. The value of k for the non-periodic dimension(s) is set by fill_values
- cartesian – if True, the coordinates given in input are treated as in cartesian units. If False, the coordinates are crystal, i.e. in units of b1,b2,b3. Default = False
- labels – optional, the list of labels to be set for some of the kpoints. See labels for more info
- weights – optional, a list of floats with the weight associated to the kpoint list
- fill_values – scalar to be set to all non-periodic dimensions (indicated by False in self.pbc), or list of values for each of the non-periodic dimensions.
- kpoints –
-
set_kpoints_mesh(mesh, offset=[0.0, 0.0, 0.0])¶ Set KpointsData to represent a uniformily spaced mesh of kpoints in the Brillouin zone. This excludes the possibility of set/get kpoints
Parameters: - mesh – a list of three integers, representing the size of the kpoint mesh along b1,b2,b3.
- offset – (optional) a list of three floats between 0 and 1. [0.,0.,0.] is Gamma centered mesh [0.5,0.5,0.5] is half shifted [1.,1.,1.] by periodicity should be equivalent to [0.,0.,0.] Default = [0.,0.,0.].
-
set_kpoints_mesh_from_density(distance, offset=[0.0, 0.0, 0.0], force_parity=False)¶ Set a kpoints mesh using a kpoints density, expressed as the maximum distance between adjacent points along a reciprocal axis
Parameters: - distance – distance (in 1/Angstrom) between adjacent
kpoints, i.e. the number of kpoints along each reciprocal
axis i is
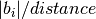 where
where 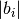 is the norm of the reciprocal cell vector.
is the norm of the reciprocal cell vector. - offset – (optional) a list of three floats between 0 and 1. [0.,0.,0.] is Gamma centered mesh [0.5,0.5,0.5] is half shifted Default = [0.,0.,0.].
- force_parity – (optional) if True, force each integer in the mesh to be even (except for the non-periodic directions).
Note: a cell should be defined first.
Note: the number of kpoints along non-periodic axes is always 1.
- distance – distance (in 1/Angstrom) between adjacent
kpoints, i.e. the number of kpoints along each reciprocal
axis i is
-
set_kpoints_path(value=None, kpoint_distance=None, cartesian=False, epsilon_length=1e-05, epsilon_angle=1e-05)¶ Set a path of kpoints in the Brillouin zone.
Deprecated since version 0.11: Use the methods inside the aiida.tools.data.array.kpoints module instead.
Parameters: - value –
description of the path, in various possible formats.
None: automatically sets all irreducible high symmetry paths. Requires that a cell was set
or
[(‘G’,’M’), (…), …] [(‘G’,’M’,30), (…), …] [(‘G’,(0,0,0),’M’,(1,1,1)), (…), …] [(‘G’,(0,0,0),’M’,(1,1,1),30), (…), …]
- cartesian (bool) – if set to true, reads the coordinates eventually passed in value as cartesian coordinates. Default: False.
- kpoint_distance (float) – parameter controlling the distance between kpoints. Distance is given in crystal coordinates, i.e. the distance is computed in the space of b1,b2,b3. The distance set will be the closest possible to this value, compatible with the requirement of putting equispaced points between two special points (since extrema are included).
- epsilon_length (float) – threshold on lengths comparison, used to get the bravais lattice info. It has to be used if the user wants to be sure the right symmetries are recognized.
- epsilon_angle (float) – threshold on angles comparison, used to get the bravais lattice info. It has to be used if the user wants to be sure the right symmetries are recognized.
- value –
-
-
__label__= 'kpoints'¶
-
__module__= 'aiida.restapi.translator.data.kpoints'¶
-
_aiida_class¶
-
_aiida_type= 'data.array.kpoints.KpointsData'¶
-
_qb_type= 'data.array.kpoints.KpointsData.'¶
-
_result_type= 'kpoints'¶
-
class
-
class
aiida.restapi.translator.data.structure.StructureDataTranslator(**kwargs)[source]¶ Bases:
aiida.restapi.translator.data.DataTranslatorTranslator relative to resource ‘structures’ and aiida class StructureData
-
class
StructureData(**kwargs)¶ Bases:
aiida.orm.data.DataThis class contains the information about a given structure, i.e. a collection of sites together with a cell, the boundary conditions (whether they are periodic or not) and other related useful information.
-
__abstractmethods__= frozenset([])¶
-
__module__= 'aiida.orm.data.structure'¶
-
_abc_cache= <_weakrefset.WeakSet object>¶
-
_abc_negative_cache= <_weakrefset.WeakSet object>¶
-
_abc_negative_cache_version= 94¶
-
_abc_registry= <_weakrefset.WeakSet object>¶
-
_adjust_default_cell(vacuum_factor=1.0, vacuum_addition=10.0, pbc=(False, False, False))¶ If the structure was imported from an xyz file, it lacks a defined cell, and the default cell is taken ([[1,0,0], [0,1,0], [0,0,1]]), leading to an unphysical definition of the structure. This method will adjust the cell
-
_dimensionality_label= {0: '', 1: 'length', 2: 'surface', 3: 'volume'}¶
-
_get_cif(converter='ase', store=False, **kwargs)¶ Creates
aiida.orm.data.cif.CifData.Parameters: - converter – specify the converter. Default ‘ase’.
- store – If True, intermediate calculation gets stored in the AiiDA database for record. Default False.
Returns:
-
_get_object_ase()¶ Converts
StructureDatato ase.AtomsReturns: an ase.Atoms object
-
_get_object_phonopyatoms()¶ Converts StructureData to PhonopyAtoms
Returns: a PhonopyAtoms object
-
_get_object_pymatgen(**kwargs)¶ Converts
StructureDatato pymatgen objectReturns: a pymatgen Structure for structures with periodic boundary conditions (in three dimensions) and Molecule otherwise Note
Requires the pymatgen module (version >= 3.0.13, usage of earlier versions may cause errors).
-
_get_object_pymatgen_molecule(**kwargs)¶ Converts
StructureDatato pymatgen Molecule objectReturns: a pymatgen Molecule object corresponding to this StructureDataobject.Note
Requires the pymatgen module (version >= 3.0.13, usage of earlier versions may cause errors)
-
_get_object_pymatgen_structure(**kwargs)¶ Converts
StructureDatato pymatgen Structure object :param add_spin: True to add the spins to the pymatgen structure. Default is False (no spin added).Note
The spins are set according to the following rule:
- if the kind name ends with 1 -> spin=+1
- if the kind name ends with 2 -> spin=-1
Returns: a pymatgen Structure object corresponding to this StructureDataobjectRaises: ValueError – if periodic boundary conditions does not hold in at least one dimension of real space; if there are partial occupancies together with spins (defined by kind names ending with ‘1’ or ‘2’). Note
Requires the pymatgen module (version >= 3.0.13, usage of earlier versions may cause errors)
-
_logger= <celery.utils.log.ProcessAwareLogger object>¶
-
_parse_xyz(inputstring)¶ Read the structure from a string of format XYZ.
-
_plugin_type_string= 'data.structure.StructureData.'¶
-
_prepare_cif(main_file_name='')¶ Write the given structure to a string of format CIF.
-
_prepare_tcod(main_file_name='', **kwargs)¶ Write the given structure to a string of format TCOD CIF.
-
_prepare_xsf(main_file_name='')¶ Write the given structure to a string of format XSF (for XCrySDen).
-
_prepare_xyz(main_file_name='')¶ Write the given structure to a string of format XYZ.
-
_query_type_string= 'data.structure.'¶
-
_set_defaults¶
-
_set_incompatibilities= [('ase', 'cell'), ('ase', 'pbc'), ('ase', 'pymatgen'), ('ase', 'pymatgen_molecule'), ('ase', 'pymatgen_structure'), ('cell', 'pymatgen'), ('cell', 'pymatgen_molecule'), ('cell', 'pymatgen_structure'), ('pbc', 'pymatgen'), ('pbc', 'pymatgen_molecule'), ('pbc', 'pymatgen_structure'), ('pymatgen', 'pymatgen_molecule'), ('pymatgen', 'pymatgen_structure'), ('pymatgen_molecule', 'pymatgen_structure')]¶
-
_validate()¶ Performs some standard validation tests.
-
append_atom(**kwargs)¶ Append an atom to the Structure, taking care of creating the corresponding kind.
Parameters: - ase – the ase Atom object from which we want to create a new atom (if present, this must be the only parameter)
- position – the position of the atom (three numbers in angstrom)
- symbols – passed to the constructor of the Kind object.
- weights – passed to the constructor of the Kind object.
- name – passed to the constructor of the Kind object. See also the note below.
Note
Note on the ‘name’ parameter (that is, the name of the kind):
- if specified, no checks are done on existing species. Simply, a new kind with that name is created. If there is a name clash, a check is done: if the kinds are identical, no error is issued; otherwise, an error is issued because you are trying to store two different kinds with the same name.
- if not specified, the name is automatically generated. Before adding the kind, a check is done. If other species with the same properties already exist, no new kinds are created, but the site is added to the existing (identical) kind. (Actually, the first kind that is encountered). Otherwise, the name is made unique first, by adding to the string containing the list of chemical symbols a number starting from 1, until an unique name is found
Note
checks of equality of species are done using the
compare_with()method.
-
append_kind(kind)¶ Append a kind to the
StructureData. It makes a copy of the kind.Parameters: kind – the site to append, must be a Kind object.
-
append_site(site)¶ Append a site to the
StructureData. It makes a copy of the site.Parameters: site – the site to append. It must be a Site object.
-
cell¶ Returns the cell shape.
Returns: a 3x3 list of lists.
-
cell_angles¶ Get the angles between the cell lattice vectors in degrees.
-
cell_lengths¶ Get the lengths of cell lattice vectors in angstroms.
-
clear_kinds()¶ Removes all kinds for the StructureData object.
Note
Also clear all sites!
-
clear_sites()¶ Removes all sites for the StructureData object.
-
get_ase()¶ Get the ASE object. Requires to be able to import ase.
Returns: an ASE object corresponding to this StructureDataobject.Note
If any site is an alloy or has vacancies, a ValueError is raised (from the site.get_ase() routine).
-
get_cell_volume()¶ Returns the cell volume in Angstrom^3.
Returns: a float.
-
get_composition()¶ Returns the chemical composition of this structure as a dictionary, where each key is the kind symbol (e.g. H, Li, Ba), and each value is the number of occurences of that element in this structure. For BaZrO3 it would return {‘Ba’:1, ‘Zr’:1, ‘O’:3}. No reduction with smallest common divisor!
Returns: a dictionary with the composition
-
get_desc()¶ Returns a string with infos retrieved from StructureData node’s properties :param self: the StructureData node :return: retsrt: the description string
-
get_dimensionality()¶ This function checks the dimensionality of the structure and calculates its length/surface/volume :return: returns the dimensionality and length/surface/volume
-
get_formula(mode='hill', separator='')¶ Return a string with the chemical formula.
Parameters: - mode –
a string to specify how to generate the formula, can assume one of the following values:
- ’hill’ (default): count the number of atoms of each species,
then use Hill notation, i.e. alphabetical order with C and H
first if one or several C atom(s) is (are) present, e.g.
['C','H','H','H','O','C','H','H','H']will return'C2H6O'['S','O','O','H','O','H','O']will return'H2O4S'From E. A. Hill, J. Am. Chem. Soc., 22 (8), pp 478–494 (1900) - ’hill_compact’: same as hill but the number of atoms for each
species is divided by the greatest common divisor of all of them, e.g.
['C','H','H','H','O','C','H','H','H','O','O','O']will return'CH3O2' - ’reduce’: group repeated symbols e.g.
['Ba', 'Ti', 'O', 'O', 'O', 'Ba', 'Ti', 'O', 'O', 'O', 'Ba', 'Ti', 'Ti', 'O', 'O', 'O']will return'BaTiO3BaTiO3BaTi2O3' - ’group’: will try to group as much as possible parts of the formula
e.g.
['Ba', 'Ti', 'O', 'O', 'O', 'Ba', 'Ti', 'O', 'O', 'O', 'Ba', 'Ti', 'Ti', 'O', 'O', 'O']will return'(BaTiO3)2BaTi2O3' - ’count’: same as hill (i.e. one just counts the number
of atoms of each species) without the re-ordering (take the
order of the atomic sites), e.g.
['Ba', 'Ti', 'O', 'O', 'O','Ba', 'Ti', 'O', 'O', 'O']will return'Ba2Ti2O6' - ’count_compact’: same as count but the number of atoms
for each species is divided by the greatest common divisor of
all of them, e.g.
['Ba', 'Ti', 'O', 'O', 'O','Ba', 'Ti', 'O', 'O', 'O']will return'BaTiO3'
- ’hill’ (default): count the number of atoms of each species,
then use Hill notation, i.e. alphabetical order with C and H
first if one or several C atom(s) is (are) present, e.g.
- separator – a string used to concatenate symbols. Default empty.
Returns: a string with the formula
Note
in modes reduce, group, count and count_compact, the initial order in which the atoms were appended by the user is used to group and/or order the symbols in the formula
- mode –
-
get_kind(kind_name)¶ Return the kind object associated with the given kind name.
Parameters: kind_name – String, the name of the kind you want to get Returns: The Kind object associated with the given kind_name, if a Kind with the given name is present in the structure. Raise: ValueError if the kind_name is not present.
-
get_kind_names()¶ Return a list of kind names (in the same order of the
self.kindsproperty, but return the names rather than Kind objects)Note
This is NOT necessarily a list of chemical symbols! Use get_symbols_set for chemical symbols
Returns: a list of strings.
-
get_pymatgen(**kwargs)¶ Get pymatgen object. Returns Structure for structures with periodic boundary conditions (in three dimensions) and Molecule otherwise. :param add_spin: True to add the spins to the pymatgen structure. Default is False (no spin added).
Note
The spins are set according to the following rule:
- if the kind name ends with 1 -> spin=+1
- if the kind name ends with 2 -> spin=-1
Note
Requires the pymatgen module (version >= 3.0.13, usage of earlier versions may cause errors).
-
get_pymatgen_molecule()¶ Get the pymatgen Molecule object.
Note
Requires the pymatgen module (version >= 3.0.13, usage of earlier versions may cause errors).
Returns: a pymatgen Molecule object corresponding to this StructureDataobject.
-
get_pymatgen_structure(**kwargs)¶ Get the pymatgen Structure object. :param add_spin: True to add the spins to the pymatgen structure. Default is False (no spin added).
Note
The spins are set according to the following rule:
- if the kind name ends with 1 -> spin=+1
- if the kind name ends with 2 -> spin=-1
Note
Requires the pymatgen module (version >= 3.0.13, usage of earlier versions may cause errors).
Returns: a pymatgen Structure object corresponding to this StructureDataobject.Raises: ValueError – if periodic boundary conditions do not hold in at least one dimension of real space.
-
get_site_kindnames()¶ Return a list with length equal to the number of sites of this structure, where each element of the list is the kind name of the corresponding site.
Note
This is NOT necessarily a list of chemical symbols! Use
[ self.get_kind(s.kind_name).get_symbols_string() for s in self.sites]for chemical symbolsReturns: a list of strings
-
get_symbols_set()¶ Return a set containing the names of all elements involved in this structure (i.e., for it joins the list of symbols for each kind k in the structure).
Returns: a set of strings of element names.
-
has_vacancies()¶ To understand if there are vacancies in the structure.
Returns: a boolean, True if at least one kind has a vacancy
-
is_alloy()¶ To understand if there are alloys in the structure.
Returns: a boolean, True if at least one kind is an alloy
-
kinds¶ Returns a list of kinds.
-
pbc¶ Get the periodic boundary conditions.
Returns: a tuple of three booleans, each one tells if there are periodic boundary conditions for the i-th real-space direction (i=1,2,3)
-
reset_cell(new_cell)¶ Reset the cell of a structure not yet stored to a new value.
Parameters: new_cell – list specifying the cell vectors Raises: ModificationNotAllowed: if object is already stored
-
reset_sites_positions(new_positions, conserve_particle=True)¶ Replace all the Site positions attached to the Structure
Parameters: - new_positions – list of (3D) positions for every sites.
- conserve_particle – if True, allows the possibility of removing a site. currently not implemented.
Raises: - ModificationNotAllowed – if object is stored already
- ValueError – if positions are invalid
Note
it is assumed that the order of the new_positions is given in the same order of the one it’s substituting, i.e. the kind of the site will not be checked.
-
set_ase(aseatoms)¶ Load the structure from a ASE object
-
set_cell(value)¶
-
set_cell_angles(value)¶
-
set_cell_lengths(value)¶
-
set_pbc(value)¶
-
set_pymatgen(obj, **kwargs)¶ Load the structure from a pymatgen object.
Note
Requires the pymatgen module (version >= 3.0.13, usage of earlier versions may cause errors).
-
set_pymatgen_molecule(mol, margin=5)¶ Load the structure from a pymatgen Molecule object.
Parameters: margin – the margin to be added in all directions of the bounding box of the molecule. Note
Requires the pymatgen module (version >= 3.0.13, usage of earlier versions may cause errors).
-
set_pymatgen_structure(struct)¶ Load the structure from a pymatgen Structure object.
Note
periodic boundary conditions are set to True in all three directions.
Note
Requires the pymatgen module (version >= 3.3.5, usage of earlier versions may cause errors).
Raises: ValueError – if there are partial occupancies together with spins.
-
sites¶ Returns a list of sites.
-
-
__label__= 'structures'¶
-
__module__= 'aiida.restapi.translator.data.structure'¶
-
_aiida_class¶
-
_aiida_type= 'data.structure.StructureData'¶
-
_qb_type= 'data.structure.StructureData.'¶
-
_result_type= 'structures'¶
-
class
-
aiida.restapi.translator.data.structure.atom_kinds_to_html(atom_kind)[source]¶ Construct in html format
an alloy with 0.5 Ge, 0.4 Si and 0.1 vacancy is represented as Ge<sub>0.5</sub> + Si<sub>0.4</sub> + vacancy<sub>0.1</sub>
- Args:
- atom_kind: a string with the name of the atomic kind, as printed by kind.get_symbols_string(), e.g. Ba0.80Ca0.10X0.10
- Returns:
- html code for rendered formula
-
class
aiida.restapi.translator.data.upf.UpfDataTranslator(**kwargs)[source]¶ Bases:
aiida.restapi.translator.data.DataTranslatorTranslator relative to resource ‘upfs’ and aiida class UpfData
-
class
UpfData(**kwargs)¶ Bases:
aiida.orm.data.singlefile.SinglefileDataFunction not yet documented.
-
__abstractmethods__= frozenset([])¶
-
__module__= 'aiida.orm.data.upf'¶
-
_abc_cache= <_weakrefset.WeakSet object>¶
-
_abc_negative_cache= <_weakrefset.WeakSet object>¶
-
_abc_negative_cache_version= 119¶
-
_abc_registry= <_weakrefset.WeakSet object>¶
-
_logger= <celery.utils.log.ProcessAwareLogger object>¶
-
_plugin_type_string= 'data.upf.UpfData.'¶
-
_query_type_string= 'data.upf.'¶
-
_validate()¶ Perform validation of the Data object.
Note
validation of data source checks license and requires attribution to be provided in field ‘description’ of source in the case of any CC-BY* license. If such requirement is too strict, one can remove/comment it out.
-
element¶
-
classmethod
from_md5(md5)¶ Return a list of all UPF pseudopotentials that match a given MD5 hash.
Note that the hash has to be stored in a _md5 attribute, otherwise the pseudo will not be found.
-
classmethod
get_or_create(filename, use_first=False, store_upf=True)¶ Pass the same parameter of the init; if a file with the same md5 is found, that UpfData is returned.
Parameters: - filename – an absolute filename on disk
- use_first – if False (default), raise an exception if more than one potential is found. If it is True, instead, use the first available pseudopotential.
- store_upf (bool) – If false, the UpfData objects are not stored in the database. default=True.
Return (upf, created): where upf is the UpfData object, and create is either True if the object was created, or False if the object was retrieved from the DB.
-
get_upf_family_names()¶ Get the list of all upf family names to which the pseudo belongs
-
classmethod
get_upf_group(group_name)¶ Return the UpfFamily group with the given name.
-
classmethod
get_upf_groups(filter_elements=None, user=None)¶ Return all names of groups of type UpfFamily, possibly with some filters.
Parameters: - filter_elements – A string or a list of strings. If present, returns only the groups that contains one Upf for every element present in the list. Default=None, meaning that all families are returned.
- user – if None (default), return the groups for all users. If defined, it should be either a DbUser instance, or a string for the username (that is, the user email).
-
md5sum¶
-
set_file(filename)¶ I pre-parse the file to store the attributes.
-
store(*args, **kwargs)¶ Store the node, reparsing the file so that the md5 and the element are correctly reset.
-
upffamily_type_string= 'data.upf.family'¶
-
-
__label__= 'upfs'¶
-
__module__= 'aiida.restapi.translator.data.upf'¶
-
_aiida_class¶ alias of
aiida.orm.data.upf.UpfData
-
_aiida_type= 'data.upf.UpfData'¶
-
_qb_type= 'data.upf.UpfData.'¶
-
_result_type= 'upfs'¶
-
class