Projwfc¶
Description¶
Use the plugin to support inputs of Quantum Espresso projwfc.x executable. Computes the
projections of atomic orbitals, 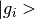 onto bloch wavefunctions
onto bloch wavefunctions 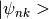 , that is
, that is
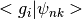 .
Also computes the DOS as a function of energy, and the PDOS for each . The orbitals
are found using the PseudoPotential used for the PW calculation.
See the projwfc doc see the QE documentation for more details.
.
Also computes the DOS as a function of energy, and the PDOS for each . The orbitals
are found using the PseudoPotential used for the PW calculation.
See the projwfc doc see the QE documentation for more details.
Supported codes¶
- tested from projwfc.x v.5.1.2 onwards
Inputs¶
- parent_calculation, A PW calculation. It is also recommended that a bands calculation be used as the parent for the best viewing results, though this is not mandatory.
- parameters, class
ParameterDataInput parameters of projwfc.x, as a nested dictionary, mapping the input of QE. See the QE documentation for the full list of variables and their meaning.
Outputs¶
There are several output nodes that can be created by the plugin.
All output nodes can be accessed with the calculation.out method.
output_parameters
ParameterDataContains the wall time of the run, as well as any warnings that may occurred.projections
ProjectionDataContains the projections which store the orbitals, pdos arrays, and projection arrays.orbitals
RealhydrogenOrbitalwhich can be called using:projection.get_orbitals()
are essentially collections of dictionaries whose elements describe a real-space hydrogen-like orbital descriptors as keys. These keys, and their values, can be found using orbital.get_orbital_dict(). For example running the following commands on the output of a projwfc output for BaTiO3:
projection = my_projwfc_calc.out.projections # the projection data this_orbital = projection.get_orbitals()[0] # first element in a list of orbitals this_orbital.get_orbital_dict() # retrieves the orbital dictionary
Would have typical output such as the following:
{'angular_momentum': 0, 'diffusivity': None, 'kind_name': u'Ba', 'magnetic_number': 0, 'module_name': 'realhydrogen', 'position': [0.0, 0.0, 0.0], 'radial_nodes': 0, 'spin': None, 'spin_orientation': None, 'x_orientation': None, 'z_orientation': None}
Where this particular orbital would be the one associated with an ‘1S’ orbital (see angular_momentum, magnetic_number and radial_nodes) centered at [0.0,0.0,0.0] (position) in the cell which is where a Ba atom lies (kind_name). Note that many descriptors have the value of None, meaning the plugin did not attempt to find associated values for these parameters, this is normal. You can call specific orbitals by passing dictionary keys during the
get_orbitals()command. For example:projection.get_orbitals()
will call all of the orbitals stored. But if we only wanted to recall all of the ‘P’ orbitals on any oxygen we could use:
projection.get_orbitals(kind_name='O',angular_momentum=1)
or:
this_dict = {'kind_name':'O','angular_momentum':1} projection.get_orbitals(**this_dict)
Note
If you want to quickly find what angular_momentum and magnetic_number is associated with which common orbital name you can use the convenience method
get_quantum_numbers_from_nameprojections, arrays showing the
projections where will be associated with a specific orbital and are the bloch waves.
They can be called using:projection.get_projections(**this_dict)
Where
this_dictcan be a dictionary to retrieve specific projections, with the exact same syntax described earlier for orbitals. Typical output would be:[(orbital_1, projectionarray_1), (orbital_2, projectionarray_2),...]
Note
In the case where spin-polarized calculations are used in the parent, there will be two output projections. One each for spin up and spin down.
pdos, arrays showing the pdos for a given orbital,
Again, this uses the same orbital dictionary syntax described in orbitals. Typical output
would be:[(orbital_1, energyarray_1, pdosarray_1), (orbital_2, energyarray_2, pdosarray_2),...]
where the pdosarrays show the projected density of state for a given orbital using the energyarrays as their ‘axis’
- bands
BandsDataParsed energy for each band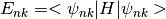 . The projections output will have a reference to the bands accessible using
. The projections output will have a reference to the bands accessible using projection.get_reference_bandsdata()
Note
In the case where spin-polarized calculations are used in the parent, there will be two output bands. One each for spin up and spin down.
Dos
XyDataContains the absolute Dos, which should not be confused with the sum of all the pdos. The energy axis and dos can be found using:Dos.get_x() Dos.get_y()
Which will return the tuples (in order):
(u'Energy', Energy_array, 'eV') (u'Dos', Dos_array, 'states/eV')
Where the Energy_array is a numpy array given the energy values and the Dos_array is a numpy array giving the Dos values for each energy in the Energy_array.
Errors¶
Errors of the parsing are reported in the log of the calculation (accessible
with the verdi calculation logshow command).
Moreover, they are stored in the ParameterData under the key warnings, and are
accessible with Calculation.res.warnings.